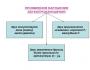
Shembuj të llojit dominues të trashëgimisë X të lidhur. Trashëgimia recesive e lidhur me X. Pedigree me mbizotërues të lidhur me X
E natyrshme në disa forma të patologjisë, për shembull, vitamina
D-rakit. Të dy homozigotët dhe heterozigotët do të kenë një manifestim fenotipik të sëmundjes. Martesat e ndryshme janë gjenetikisht të mundshme, por ato në të cilat babai është i sëmurë janë informuese. Në një martesë me një grua të shëndetshme, vërehen tiparet e mëposhtme të trashëgimisë së patologjive:
1) të gjithë djemtë dhe fëmijët e tyre do të jenë të shëndetshëm, pasi vetëm kromozomi Y mund t'u kalohet atyre nga babai i tyre;
2) të gjitha vajzat do të jenë heterozigote dhe fenotipisht të sëmura.
Këto dy karakteristika e dallojnë këtë lloj nga tipi autosomik dominant, në të cilin raporti i vëllezërve e motrave të sëmurë dhe të shëndetshëm është 1:1 dhe janë njësoj të padallueshëm për fëmijët nga ata me një model të trashëgimisë autosomale dominuese (1:1), dhe gjithashtu duhet të ketë nuk ka dallime gjinore. Ka një manifestim më të fortë të sëmundjes tek meshkujt, pasi ata nuk kanë efektin kompensues të rrugicës normale. Literatura përshkruan origjinën e disa sëmundjeve me këtë lloj transmetimi, të cilat nuk kanë vëllezër e motra meshkuj, pasi shkalla e rëndë e dëmtimit shkakton vdekjen e tyre intrauterine. Kjo origjinë duket e veçantë: pasardhësit janë vetëm femra, rreth gjysma e tyre janë të sëmurë dhe anamneza mund të përfshijë aborte spontane dhe lindje të fetuseve mashkullore.
Llojet e listuara të trashëgimisë përfshijnë kryesisht sëmundje monogjenike (të përcaktuara nga një mutacion i një gjeni). Megjithatë, gjendja patologjike mund të varet nga dy ose më shumë gjene mutant. Një sërë gjenesh patologjike kanë reduktuar penetrencën. Për më tepër, prania e tyre në gjenom, edhe në gjendje homozigote, është e nevojshme, por jo e mjaftueshme për zhvillimin e sëmundjes. Kështu, jo të gjitha llojet e trashëgimisë së sëmundjeve njerëzore përshtaten në tre skemat e listuara më sipër.
METODAT PËR PËRCAKTIMIN E DEFEKTIT BIOKIMIK PRIMAR.
Kur shqyrtohet historia e zbulimit të formave nozologjike monogjenike, shihet qartë se periudha më e gjatë e saj, afërsisht deri në mesin e viteve 50, lidhet me identifikimin e formave të tilla në bazë të një ekzaminimi klinik dhe gjenealogjik të familjeve. Megjithatë, kjo periudhë nuk është shumë produktive. Për shembull, 18 format gjenetike të identifikuara aktualisht të mukopolisakaridozave trashëgimore, të shkaktuara nga mutacionet e 11-12 gjeneve të ndryshme, formojnë klinikisht vetëm dy fenotipe paksa të ndryshme, dhe bazuar në pamjen klinike dhe llojin e trashëgimisë, janë zbuluar vetëm dy njësi nozologjike - Sindroma Hurler dhe sindroma Hunter. E njëjta situatë është zhvilluar me klasat e tjera të defekteve metabolike trashëgimore. Zbulimi dhe përshkrimi i sëmundjeve trashëgimore nuk duhet të konsiderohet i plotë. Aktualisht njihen rreth dy mijë gjendje patologjike Mendeliane. Teorikisht, bazuar në numrin e përgjithshëm të gjeneve strukturore të rendit prej 50-100 mijë, mund të supozohet se shumica e aleleve mutant patologjike nuk janë zbuluar ende. Edhe nëse pranojmë se shumë mutacione të tilla janë vdekjeprurëse, ndërsa të tjerët, përkundrazi, nuk ndikojnë në funksione serioze dhe nuk njihen klinikisht, atëherë duhet të presim zbulimin e vazhdueshëm të gjithnjë e më shumë formave të reja të patologjisë trashëgimore. Por mund të themi me besim se sëmundjet më të zakonshme që japin një pamje të qartë klinike tashmë janë përshkruar. Format e reja të zbuluara janë rezultat i mutacioneve të rralla. Përveç kësaj, nga pikëpamja gjenetike, do të rezultojnë mutacione të të njëjtit gjen, por që prekin struktura të reja ose duke qenë të ndryshëm në natyrën e tyre molekulare (për shembull, mutacione në pjesën rregullatore dhe jo strukturore të gjenit). Kjo është arsyeja pse zbulimi i aleleve të reja mutante dhe copëzimi i sëmundjeve të njohura në forma gjenetikisht të ndryshme janë të pandashme nga lidhja me analizën gjenetike klinike tradicionale të qasjeve të reja gjenetike që bëjnë të mundur arritjen e tipareve elementare më diskrete dhe më afruese.
Vendin e parë e zënë metodat biokimike. Qasja biokimike u aplikua për herë të parë dhe doli të ishte shumë e frytshme në fillim të këtij shekulli në studimin klinik dhe gjenetik të alkaptunurisë. Pikërisht si rezultat i këtij studimi u gjet një tipar biokimik mendelian për një nga sëmundjet trashëgimore, në formën e sekretimit të tepërt të acidit homogjentisik në urinë, dhe u sugjerua se ka sëmundje të ngjashme të lindura metabolike me specifikat e tyre. defekt biokimik. Aktualisht, më shumë se 300 sëmundje metabolike trashëgimore me anomali të studiuara janë përshkruar në gjenetikën biokimike. Në praktikën klinike, për diagnozën biokimike të sëmundjeve të njohura metabolike, përdoret një sistem testesh cilësore dhe gjysmë sasiore, me ndihmën e të cilave është e mundur të zbulohet përmbajtja e dëmtuar e produkteve metabolike (për shembull, sekretimi i tepërt urinar i fenilpiruvicit. acid në fenilketonuri ose homocistinë në homocistinuri). Përdorimi i llojeve të ndryshme të elektroforezës dhe kromatografisë veç e veç dhe në kombinim, si dhe metoda të tjera, bën të mundur përcaktimin se cila lidhje metabolike është e prishur. Për të zbuluar se cila enzimë ose proteinë tjetër është e përfshirë në efektin metabolik dhe cili është ndryshimi i proteinave, si rregull, përdoren jo vetëm lëngjet biologjike, por edhe qelizat e pacientit, dhe përdoren metoda komplekse për të përcaktuar përmbajtjen e enzima, aktiviteti i saj katalitik dhe struktura molekulare.
Metodat biokimike plotësohen me metoda gjenetike molekulare, të cilat kanë rëndësi të pavarur për deshifrimin e natyrës së mutacioneve drejtpërdrejt në ADN. Tradicionalisht, përdorimi i tyre është i mundur pas identifikimit të një defekti në produktin e gjenit përkatës, por deri më tani është realist për disa raste të patologjisë, për shembull, për mutacione të gjeneve të globinës.
Frytshmëria e metodave të kërkimit biokimik është kryesisht për shkak të faktit se analiza biokimike e lëngjeve biologjike plotësohet nga analiza e qelizave të trupit. Analiza biokimike gjenetike e qelizave doli të jetë vendimtare në kalimin në diagnostikimin biokimik me analizën e metabolitëve në studimin e enzimave dhe proteinave strukturore drejtpërdrejt, në veçanti receptorët qelizorë.
Kjo çoi në zbulimin e defekteve parësore në molekulat e proteinave dhe shumë sëmundje trashëgimore. Metodat imunologjike janë të afërta në aftësitë e tyre me metodat biokimike. Diagnostifikimi dhe studimi i thelluar i formave gjenetike të kushteve të ndryshme të mungesës së imunitetit të trashëguar bazohen në metodat për vlerësimin e nivelit të imunoglobulinave serike të klasave të ndryshme, si dhe gjendjen e imunitetit qelizor. Një vend të spikatur në arsenalin e këtyre metodave zënë reaksionet serologjike klasike me eritrocite ose leukocite për të përcaktuar statusin e antigjeneve sipërfaqësore. Vitet e fundit janë përdorur gjithnjë e më shumë metoda radioimunokimike për përcaktimin e defektit të hormoneve dhe disa substancave të tjera biologjikisht aktive.
Të gjitha këto metoda përdoren për të identifikuar defektet biokimike dhe natyrën molekulare të mutacioneve me një përqasje popullate-gjeografike. Rëndësia e kësaj qasjeje është se defekte dhe mutacione të rralla mund të ndodhin kryesisht në rajone të caktuara gjeografike për shkak të kushteve specifike të mjedisit njerëzor. Mjafton të kujtojmë shpërndarjen mbizotëruese të gjenoglobinopative të ndryshme, veçanërisht në zonat ku malaria është e përhapur. Popullatat e izoluara me një numër të madh martesash fisnike shpesh shërbyen si burim për zbulimin e mutacioneve të reja për shkak të ndarjes më të shpeshtë të homozigotëve në gjendje recesive. Qasja popullatë-gjeografike gjithashtu ndihmon, me mostra të mëdha pacientësh, për të diferencuar më shpejt mutacionet fenotipike të ngjashme, por gjenetikisht të ndryshme.
Trashëgimia recesive e lidhur me X(anglisht) Trashëgimia recesive e lidhur me X ) është një nga llojet e trashëgimisë së lidhur me seksin. Një trashëgimi e tillë është tipike për tiparet gjenet e të cilave ndodhen në kromozomin X dhe që shfaqen vetëm në një gjendje homozigote ose hemizigote. Kjo lloj trashëgimie ka një sërë sëmundjesh të lindura trashëgimore tek njerëzit, këto sëmundje shoqërohen me një defekt në ndonjë nga gjenet e vendosura në kromozomin X seksual dhe shfaqen nëse nuk ka një kromozom tjetër X me një kopje normale të të njëjtit gjen. Në literaturë ekziston një shkurtim XR për të treguar trashëgiminë recesive të lidhur me X.
Është tipike për sëmundjet recesive të lidhura me X që meshkujt zakonisht preken për sëmundjet e rralla të lidhura me X, kjo është pothuajse gjithmonë e vërtetë. Të gjitha vajzat e tyre fenotipike të shëndetshme janë bartëse heterozigote. Në mesin e djemve të nënave heterozigote, raporti i të sëmurëve me të shëndetshëm është 1 me 1.
Një rast i veçantë i trashëgimisë recesive të lidhur me X është të kryqëzuar trashëgimi (anglisht) trashëgimi e kryqëzuar, Gjithashtu trashëgimi e kryqëzuar), si rezultat i së cilës karakteristikat e baballarëve shfaqen tek vajzat dhe karakteristikat e nënës tek djemtë. Ky lloj trashëgimie u emërua nga një nga autorët e teorisë kromozomale të trashëgimisë, Thomas Hunt Morgan. Ai e përshkroi për herë të parë këtë lloj trashëgimie për tiparin e ngjyrës së syve në Drosophila në 1911. Trashëgimia e kryqëzuar ndodh kur nëna është homozigote për një tipar recesiv të lokalizuar në kromozomin X, dhe babai ka një alele dominuese të këtij gjeni në të vetmin kromozomin X. Zbulimi i këtij lloji të trashëgimisë gjatë analizës së segregacionit është një nga provat e lokalizimit të gjenit përkatës në kromozomin X.
Veçoritë e trashëgimisë së tipareve recesive të lidhura me seksin tek njerëzit
Tek njerëzit, si të gjithë gjitarët, seksi mashkull është heterogametik (XY), dhe seksi femër është homogametik (XX). Kjo do të thotë se burrat kanë vetëm një kromozom X dhe një Y, ndërsa gratë kanë dy kromozome X. Kromozomet X dhe kromozomet Y kanë rajone të vogla homologe (rajone pseudoautozomale). Trashëgimia e tipareve gjenet e të cilave ndodhen në këto rajone është e ngjashme me trashëgiminë e gjeneve autosomale dhe nuk diskutohet në këtë artikull.
Tiparet e lidhura me kromozomin X mund të jenë recesive ose dominante. Tiparet recesive nuk shfaqen te individët heterozigotë në prani të një tipari dominues. Meqenëse meshkujt kanë vetëm një kromozom X, meshkujt nuk mund të jenë heterozigotë për gjenet që gjenden në kromozomin X. Për këtë arsye, tek meshkujt ekzistojnë vetëm dy gjendje të mundshme të tiparit recesiv të lidhur me X:
- nëse ka një alel në një kromozom të vetëm X që përcakton një tipar ose çrregullim, burri e shfaq atë tipar ose çrregullim, dhe të gjitha vajzat e tij e marrin këtë alel prej tij së bashku me kromozomin X (djemtë do të marrin kromozomin Y);
- nëse nuk ka një alele të tillë në të vetmin kromozomin X, atëherë ky tipar ose çrregullim nuk manifestohet te një mashkull dhe nuk kalon te pasardhësit e tij.
Meqenëse gratë kanë dy kromozome X, ato kanë tre kushte të mundshme për tipare recesive të lidhura me X:
- aleli që përcakton këtë tipar ose çrregullim mungon në të dy kromozomet X - tipari ose çrregullimi nuk manifestohet dhe nuk transmetohet te pasardhësit;
- aleli që përcakton tiparin ose çrregullimin është i pranishëm vetëm në një kromozom X - tipari ose çrregullimi zakonisht nuk shfaqet, dhe kur trashëgohet, afërsisht 50% e pasardhësve e marrin këtë alel së bashku me kromozomin X prej tij (50% tjetër nga pasardhësit do të marrin një tjetër kromozom X);
- aleli që përcakton tiparin ose çrregullimin është i pranishëm në të dy kromozomet X - tipari ose çrregullimi manifestohet dhe kalohet te pasardhësit në 100% të rasteve.
Disa çrregullime të trashëguara në një model recesiv të lidhur me X mund të jenë aq të rënda sa të çojnë në vdekjen e fetusit. Në këtë rast, mund të mos ketë një pacient të vetëm të njohur midis anëtarëve të familjes dhe midis paraardhësve të tyre.
Gratë që kanë vetëm një kopje të mutacionit quhen bartëse. Në mënyrë tipike, një mutacion i tillë nuk shprehet në fenotip, domethënë nuk manifestohet në asnjë mënyrë. Disa sëmundje me trashëgimi recesive të lidhura me X kanë ende disa manifestime klinike në bartëset femra për shkak të mekanizmit të kompensimit të dozës, për shkak të të cilave një nga kromozomet X inaktivizohet rastësisht në qelizat somatike, dhe në disa qeliza të trupit një alele X është shprehur, dhe në të tjerët - një tjetër.
Disa sëmundje recesive të lidhura me X tek njerëzit
I zakonshëm
Sëmundjet e zakonshme recesive të lidhura me X:
- Çrregullimi i trashëguar i shikimit të ngjyrave (verbëria e ngjyrave). Në Evropën Veriore, afërsisht 8% e meshkujve dhe 0,5% e femrave vuajnë nga shkallë të ndryshme të dobësisë së perceptimit të kuq-gjelbër.
- Iktioza e lidhur me X. Njolla të thata dhe të ashpra shfaqen në lëkurën e pacientëve për shkak të akumulimit të tepërt të steroideve të sulfonuara. Ndodh në 1 në 2000-6000 meshkuj.
- Distrofia muskulare Duchenne. Një sëmundje e shoqëruar me degjenerim të indit muskulor dhe që çon në vdekje në moshë të re. Ndodh në 1 në 3600 të porsalindur meshkuj.
- Hemofilia A (hemofilia klasike). Sëmundja e lidhur me mungesën e faktorit VIII të koagulimit të gjakut shfaqet në një në 4000-5000 meshkuj.
- Hemofilia B. Një sëmundje e lidhur me mungesën e faktorit IX të koagulimit të gjakut, shfaqet në një në 20,000-25,000 burra.
- Distrofia muskulare Becker. Sëmundja është e ngjashme me distrofinë muskulare Duchenne, por është disi më e lehtë. Ndodh në 3-6 nga 100.000 të porsalindurit meshkuj.
- Sindroma Kabuki - defekte të shumta të lindjes (defekte në zemër, mangësi në rritje, humbje dëgjimi, anomali të traktit urinar) dhe prapambetje mendore. Prevalenca 1:32000.
- Sindroma e pandjeshmërisë ndaj androgjenit (sindroma Morris) - një individ me sindromë të plotë ka një pamje femërore, gjoks dhe vaginë të zhvilluar, pavarësisht nga kariotipi 46XY dhe testikujt e pazbritur. Shkalla e incidencës është nga 1:20,400 në 1:130,000 të sapolindur me një kariotip 46,XY.
I rrallë
- Sëmundja e Brutonit (agammaglobulinemia kongjenitale). Imunodefiçenca humorale primare. Ndodh te djemtë me një frekuencë prej 1:100,000 - 1:250,000.
- Sindroma Wiskott-Aldrich është një mungesë imuniteti dhe trombocitopeni kongjenitale. Prevalenca: 4 raste për 1 000 000 lindje meshkuj.
- Sindroma Lowe (sindroma okulocerebrorenale) - anomalitë e skeletit, çrregullimet e ndryshme të veshkave, glaukoma dhe kataraktet që nga fëmijëria e hershme. Ndodh me një frekuencë prej 1:500,000 të porsalindur meshkuj.
- Sindroma Allan-Herndon-Dudley është një sindromë e rrallë, e gjetur vetëm tek meshkujt, në të cilën zhvillimi i trurit pas lindjes është i dëmtuar. Sindroma shkaktohet nga një mutacion në gjenin MCT8, i cili kodon një proteinë që transporton hormonin e tiroides. Përshkruar për herë të parë në 1944.
8. Koncepti i “lidhjes” së gjeneve. Trashëgimia e tipareve të lidhura me X tek njerëzit.
Një fenomen i bazuar në lokalizimin e gjeneve në një kromozom. Lidhja e gjeneve u zbulua për herë të parë në vitin 1906 nga W. Bateson dhe R. Punnett në eksperimentet mbi kryqëzimin e bizeleve të ëmbla. Më vonë, lidhja e gjeneve u studiua në detaje nga T. Morgan dhe kolegët e tij në eksperimentet me Drosophila. Lidhja e gjeneve shprehet në faktin se alelet e gjeneve të lidhura që janë në të njëjtin grup lidhjesh priren të trashëgohen së bashku. Kjo çon në formimin e gameteve preem në hibrid. me kombinime “prindërore” të aleleve. Për të treguar lidhjen e gjeneve, përdoren simbolet AB/av ose AB/Ab lidhja e aleleve dominante (ose recesive) me njëri-tjetrin quhet AB/av. faza e lidhjes, dhe lidhja e aleleve dominante me Av/aB recesive është faza e repulsionit. Në të dyja rastet, lidhja e gjeneve rezulton në një frekuencë më të ulët të individëve me kombinime "jo-prindërore", rekombinante të tipareve sesa do të pritej nga trashëgimia e pavarur e tipareve. Me lidhjen e plotë të gjeneve, formohen vetëm dy lloje gametesh (me kombinimet origjinale të gjeneve të lidhura me lidhje jo të plotë, formohen kombinime të reja të aleleve të gjeneve të lidhura). Lidhja jo e plotë e gjeneve është rezultat i kryqëzimit midis gjeneve të lidhura, prandaj lidhja e plotë e gjeneve është e mundur në organizmat në qelizat e të cilëve kalimi normalisht nuk ndodh (për shembull, qelizat germinale të Drosophila males). Kështu, lidhja e plotë e gjeneve është më tepër një përjashtim nga rregulli i lidhjes jo të plotë të gjeneve. Përveç kësaj, lidhja e plotë e gjeneve mund të simulohet nga fenomeni i pleiotropisë. Në disa raste, në mejozë, ndodh rregullisht divergjenca jo e rastësishme e kromozomeve johomologe në një pol, gjë që çon në formimin e gameteve. me kombinime të caktuara të aleleve të gjeneve të palidhura. Çiftet e ndryshme të gjeneve brenda të njëjtit grup lidhës karakterizohen nga shkallë të ndryshme lidhjeje në varësi të distancës ndërmjet tyre. Sa më e madhe të jetë distanca midis gjeneve në një kromozom, aq më e vogël është forca e ngjitjes midis tyre dhe aq më shpesh formohen llojet rekombinante të gameteve. Studimi i lidhjes së gjeneve dhe trashëgimisë së ndërlidhur të tipareve shërbeu si një nga konfirmimet e teorisë kromozomale të trashëgimisë dhe shtysa fillestare për analizën dhe zhvillimin e teorisë së kryqëzimit.
Trashëgimia e lidhur me X
Meqenëse kromozomi X është i pranishëm në kariotipin e çdo personi, tiparet e trashëguara të lidhura me kromozomin X shfaqen tek përfaqësuesit e të dy gjinive. Gratë i marrin këto gjene nga të dy prindërit dhe ua kalojnë pasardhësve përmes gameteve të tyre. Meshkujt marrin një kromozom X nga nëna e tyre dhe ia kalojnë atë pasardhësve të tyre femra.
Ka trashëgimi dominante të lidhura me X dhe recesive të lidhura me X. Tek njerëzit, një tipar mbizotërues i lidhur me X transmetohet nga nëna tek të gjithë pasardhësit. Një burrë ua përcjell tiparin e tij mbizotërues të lidhur me X vetëm te vajzat e tij. Një tipar recesiv i lidhur me X tek gratë shfaqet vetëm nëse ato marrin alelin përkatës nga të dy prindërit. Tek meshkujt, ajo zhvillohet kur ata marrin një alele recesive nga nëna e tyre. Gratë ua kalojnë alelën recesive pasardhësve të të dy gjinive, ndërsa meshkujt vetëm vajzave të tyre.
Me trashëgiminë e lidhur me X, është i mundur një karakter i ndërmjetëm i manifestimit të tiparit te heterozigotët.
Gjenet e lidhura me Y janë të pranishme në gjenotipin e vetëm meshkujve dhe kalohen brez pas brezi nga babai te djali.
Trashëgimia e lidhur me X e një tipari recesiv nga një baba i prekur.
Trashëgimia e kryqëzuar e ngjyrës së syve në Drosophila. Të gjithë djemtë e një nëne homozigote për tiparin recesiv "sy të bardhë" kanë sy të bardhë. Të gjitha vajzat kanë sy të kuq, pasi kanë trashëguar nga babai i tyre një alel dominues që shkakton skuqjen e syve.
Trashëgimia recesive e lidhur me X(anglisht) Trashëgimia recesive e lidhur me X) është një nga llojet e trashëgimisë së lidhur me seksin. Një trashëgimi e tillë është tipike për tiparet gjenet e të cilave ndodhen në kromozomin X dhe që shfaqen vetëm në një gjendje homozigote ose hemizigote. Kjo lloj trashëgimie ka një sërë sëmundjesh të lindura trashëgimore tek njerëzit, këto sëmundje shoqërohen me një defekt në ndonjë nga gjenet e vendosura në kromozomin X seksual dhe shfaqen nëse nuk ka një kromozom tjetër X me një kopje normale të të njëjtit gjen. Në literaturë ekziston një shkurtim XR për të treguar trashëgiminë recesive të lidhur me X.
Është tipike për sëmundjet recesive të lidhura me X që meshkujt zakonisht preken për sëmundjet e rralla të lidhura me X, kjo është pothuajse gjithmonë e vërtetë. Të gjitha vajzat e tyre fenotipike të shëndetshme janë bartëse heterozigote. Në mesin e djemve të nënave heterozigote, raporti i të sëmurëve me të shëndetshëm është 1 me 1.
Një rast i veçantë i trashëgimisë recesive të lidhur me X është të kryqëzuar trashëgimi (eng. trashëgimi kryqëzuese, gjithashtu trashëgimi e kryqëzuar), si rezultat i së cilës karakteristikat e baballarëve shfaqen tek vajzat dhe karakteristikat e nënës tek djemtë. Ky lloj trashëgimie u emërua nga një nga autorët e teorisë kromozomale të trashëgimisë, Thomas Hunt Morgan. Ai e përshkroi për herë të parë këtë lloj trashëgimie për tiparin e ngjyrës së syve në Drosophila në 1911. Trashëgimia e kryqëzuar ndodh kur nëna është homozigote për një tipar recesiv të lokalizuar në kromozomin X, dhe babai ka një alele dominuese të këtij gjeni në të vetmin kromozomin X. Zbulimi i këtij lloji të trashëgimisë gjatë analizës së segregacionit është një nga provat e lokalizimit të gjenit përkatës në kromozomin X.
Veçoritë e trashëgimisë së tipareve recesive të lidhura me seksin tek njerëzit
Tek njerëzit, si të gjithë gjitarët, seksi mashkull është heterogametik (XY), dhe seksi femër është homogametik (XX). Kjo do të thotë se burrat kanë vetëm një kromozom X dhe një Y, ndërsa gratë kanë dy kromozome X. Kromozomet X dhe kromozomet Y kanë rajone të vogla homologe (rajone pseudoautozomale). Trashëgimia e tipareve gjenet e të cilave ndodhen në këto rajone është e ngjashme me trashëgiminë e gjeneve autosomale dhe nuk diskutohet në këtë artikull.
Tiparet e lidhura me kromozomin X mund të jenë recesive ose dominante. Tiparet recesive nuk shfaqen te individët heterozigotë në prani të një tipari dominues. Meqenëse meshkujt kanë vetëm një kromozom X, meshkujt nuk mund të jenë heterozigotë për gjenet që gjenden në kromozomin X. Për këtë arsye, vetëm dy gjendje të tipareve recesive të lidhura me X janë të mundshme tek meshkujt:
- nëse ka një alel në një kromozom të vetëm X që përcakton një tipar ose çrregullim, burri e shfaq atë tipar ose çrregullim, dhe të gjitha vajzat e tij e marrin këtë alel prej tij së bashku me kromozomin X (djemtë do të marrin kromozomin Y);
- nëse nuk ka një alele të tillë në të vetmin kromozomin X, atëherë ky tipar ose çrregullim nuk manifestohet te një mashkull dhe nuk kalon te pasardhësit e tij.
Meqenëse gratë kanë dy kromozome X, ato kanë tre kushte të mundshme për tipare recesive të lidhura me X:
- aleli që përcakton këtë tipar ose çrregullim mungon në të dy kromozomet X - tipari ose çrregullimi nuk manifestohet dhe nuk transmetohet te pasardhësit;
- aleli që përcakton tiparin ose çrregullimin është i pranishëm vetëm në një kromozom X - tipari ose çrregullimi zakonisht nuk shfaqet, dhe kur trashëgohet, afërsisht 50% e pasardhësve e marrin këtë alel së bashku me kromozomin X prej tij (50% tjetër nga pasardhësit do të marrin një tjetër kromozom X);
- aleli që përcakton tiparin ose çrregullimin është i pranishëm në të dy kromozomet X - tipari ose çrregullimi manifestohet dhe kalohet te pasardhësit në 100% të rasteve.
Disa çrregullime të trashëguara në një model recesiv të lidhur me X mund të jenë aq të rënda sa të çojnë në vdekjen e fetusit. Në këtë rast, mund të mos ketë një pacient të vetëm të njohur midis anëtarëve të familjes dhe midis paraardhësve të tyre.
Gratë që kanë vetëm një kopje të mutacionit quhen bartëse. Në mënyrë tipike, një mutacion i tillë nuk shprehet në fenotip, domethënë nuk manifestohet në asnjë mënyrë. Disa sëmundje me trashëgimi recesive të lidhura me X kanë ende disa manifestime klinike te bartëset femra për shkak të mekanizmit të kompensimit të dozës, për shkak të të cilave një nga kromozomet X inaktivizohet rastësisht në qelizat somatike, dhe në disa qeliza të trupit një alele X është shprehur, dhe në të tjerët - një tjetër.
Disa sëmundje recesive të lidhura me X tek njerëzit
I zakonshëm
Sëmundjet e zakonshme recesive të lidhura me X:
I rrallë
Shiko gjithashtu
Shënime
- Fondacioni Gift of Life. Trashëgimia recesive e lidhur me X
- Ndërveprimet e sëmundjes Seroquel XR (quetiapine).
- Një formë e re recesive e lidhur me X të ndjeshmërisë Mendeliane ndaj sëmundjes mykobateriale
- Ndjeshmëria mendeliane e lidhur me X ndaj sëmundjeve mykobakteriale
- Vogel F., Motulsky A. Gjenetika njerëzore në 3 vëllime. - M: Mir, 1989. - T. 1. - F. 162-164. - 312 s.
- Morgan T.H., Sturtevant A.H., Muller H.J., Bridges C.B.. - New York: Henry Holt and Company, 1915. - 262 f.
- Fjalori shpjegues anglisht-rus i termave gjenetikë. Arefiev V. A., Lisovenko L. A., Moskë: Shtëpia Botuese VNIRO, 1995.
- Shevchenko V. A., Topornina N. A., Stvolinskaya N. S. Gjenetika njerëzore: Libër shkollor. për studentët më të larta teksti shkollor ndërmarrjet. Botimi i 2-të, rev. dhe shtesë - M.: Humanit. ed. Qendra VLADOS, 2004. - 240 f.: ISBN 5-691-00477-8 me 116
- Dobyns WB, Filauro A. Trashëgimia e shumicës së tipareve të lidhura me X nuk është dominuese ose recesive, vetëm e lidhur me X. Am J Med Genet A. 30 gusht 2004; 129A(2): 136-43.
- Verbëria e ngjyrave OMIM, Seria Deutan; CBD
- Carlo Gelmetti; Caputo, Ruggero. Dermatologjia Pediatrike dhe Dermatopatologjia: Një Atlas Konciz. - T&F STM, 2002. - F. 160. - ISBN 1-84184-120-X.
- Distrofia muskulare Duchenne: Enciklopedia Mjekësore MedlinePlus (i papërcaktuar) . nlm.nih.gov. Marrë më 6 maj 2014.
- Barbara A Konkle, MD, Neil C Josephson, MD. Hemofilia A. Sinonimet: Hemofilia Klasike, Mungesa e Faktorit VIII. GeneReviews, 2000
- Barbara A Konkle, MD, Neil C Josephson, MD, Hemofilia B. Sinonimet: Sëmundja e Krishtlindjeve, Mungesa e Faktorit IX. GeneReviews, 2000
- Sindroma Kabuki (i papërcaktuar) . Referenca e Shtëpisë së Gjenetikës. Marrë më 6 maj 2014.
- Bangsbøll S., Qvist I., Lebech P. E., Lewinsky M. Sindroma e feminizimit të testikujve dhe tumoret gonadale të shoqëruara në Danimarkë (Anglisht) // Acta Obstet Gynecol Scand (anglisht) rusisht: ditar. - 1992. - Janar (vëll. 71, nr. 1). - F. 63-6. -
Kjo broshurë ofron informacion rreth asaj se çfarë është trashëgimia e lidhur me X dhe si trashëgohen sëmundjet e lidhura me X.
Cilat janë gjenet dhe kromozomet?
Trupi ynë përbëhet nga miliona qeliza. Shumica e qelizave përmbajnë një grup të plotë gjenesh. Një person ka mijëra gjene. Gjenet mund të krahasohen me udhëzimet që përdoren për të kontrolluar rritjen dhe funksionimin e koordinuar të të gjithë organizmit. Gjenet janë përgjegjës për shumë karakteristika të trupit tonë, si ngjyra e syve, grupi i gjakut ose gjatësia.
Figura 1: Gjenet, kromozomet dhe ADN-ja
Gjenet janë të vendosura në struktura të ngjashme me fije të quajtura kromozome. Normalisht, shumica e qelizave në trup përmbajnë 46 kromozome. Kromozomet na transmetohen nga prindërit tanë - 23 nga nëna dhe 23 nga babi, kështu që shpesh dukemi si prindërit tanë. Kështu, ne kemi dy grupe me 23 kromozome, ose 23 çifte kromozomesh. Për shkak se gjenet janë të vendosura në kromozome, ne trashëgojmë dy kopje të secilit gjen, një kopje nga secili prind. Kromozomet (dhe për rrjedhojë gjenet) përbëhen nga një përbërje kimike e quajtur ADN.
Figura 2: 23 çifte kromozomesh të shpërndara sipas madhësisë; Kromozomi numër 1 është më i madhi. Dy kromozomet e fundit janë kromozome seksuale.

Kromozomet (shih Figurën 2), të numëruara nga 1 deri në 22, janë të njëjta tek burrat dhe gratë. Kromozome të tilla quhen autozome. Kromozomet e çiftit të 23-të janë të ndryshëm tek femrat dhe meshkujt dhe quhen kromozome seksuale. Ekzistojnë 2 variante të kromozomeve seksuale: kromozomi X dhe kromozomi Y. Normalisht, gratë kanë dy kromozome X (XX), njëri prej tyre transmetohet nga nëna, tjetri nga babai. Normalisht, meshkujt kanë një kromozom X dhe një kromozom Y (XY), me kromozomin X të kaluar nga nëna dhe kromozomin Y nga babai. Kështu, Figura 2 tregon kromozomet e një njeriu, pasi çifti i fundit, i 23-të, përfaqësohet nga kombinimi XY.
Ndonjëherë një ndryshim (mutacion) ndodh në një kopje të një gjeni që prish funksionimin normal të gjenit. Një mutacion i tillë mund të çojë në zhvillimin e një sëmundjeje gjenetike (të trashëgueshme), pasi gjeni i ndryshuar nuk transmeton informacionin e nevojshëm në trup. Sëmundjet e lidhura me X shkaktohen nga ndryshimet në gjenet në kromozomin X.
Çfarë është trashëgimia e lidhur me X?
Kromozomi X përmban shumë nga gjenet që janë shumë të rëndësishme për rritjen dhe zhvillimin e organizmit. Kromozomi Y është shumë më i vogël dhe përmban më pak gjene. Siç dihet, gratë kanë dy kromozome X (XX), prandaj, nëse një kopje e një gjeni në kromozomin X ndryshon, atëherë kopja normale në kromozomin e dytë X mund të kompensojë funksionin e atij të ndryshuar. Në këtë rast, gruaja është zakonisht një bartëse e shëndetshme e sëmundjes së lidhur me X. Një bartës është një person që nuk ka shenja të sëmundjes, por ka një kopje të ndryshuar të gjenit. Në disa raste, gratë mund të kenë manifestime të moderuara të sëmundjes.
Meshkujt kanë një kromozom X dhe një Y, kështu që kur një kopje e një gjeni në kromozomin X është ndryshuar, nuk ka asnjë kopje normale të gjenit për të kompensuar funksionin. Kjo do të thotë që një njeri i tillë do të jetë i sëmurë. Sëmundjet që trashëgohen në mënyrën e përshkruar më sipër quhen recesive të lidhura me X. Shembuj të sëmundjeve të tilla janë hemofilia, distrofia muskulare Duchenne dhe sindroma X e brishtë.
Trashëgimia dominante e lidhur me X
Shumica e sëmundjeve të lidhura me X janë recesive, por në raste të rralla, sëmundjet e lidhura me X trashëgohen si dominuese. Kjo do të thotë se nëse një grua ka një kopje të ndryshuar dhe një kopje normale të gjenit, kjo do të mjaftojë që sëmundja të shfaqet. Nëse një mashkull trashëgon një kopje të ndryshuar të gjenit të kromozomit X, ai do të zhvillojë sëmundjen, pasi meshkujt kanë vetëm një kromozom X. Gratë e prekura kanë një shans 50% (1 në 2) për të pasur një fëmijë të prekur, dhe është e njëjta gjë për vajzat dhe djemtë. Një i sëmurë do të ketë të gjitha vajzat e tij të sëmura dhe të gjithë djemtë e tij do të jenë të shëndetshëm.
Si trashëgohen sëmundjet e lidhura me X?
Nëse një grua mbartëse ka një djalë, atëherë ajo mund t'i kalojë atij ose një kromozom X me një kopje normale të gjenit, ose një kromozom X me një kopje të ndryshuar të gjenit. Kështu, çdo djalë ka një shans 50% (1 në 2) për të trashëguar një kopje të ndryshuar të gjenit dhe për të zhvilluar sëmundjen. Në të njëjtën kohë, ekziston mundësia e njëjtë - 50% (1 në 2) - që djali të trashëgojë një kopje normale të gjenit, në këtë rast ai nuk do të ketë sëmundjen. Ky probabilitet është i njëjtë për çdo djalë (Figura 3).
Nëse një grua mbartëse ka një vajzë, ajo do të kalojë ose një kromozom X me një kopje normale të gjenit ose një kromozom X me një kopje të ndryshuar. Kështu, çdo vajzë ka një shans 50% (1 në 2) për të trashëguar një kopje të ndryshuar të gjenit, në të cilin rast ajo do të jetë bartëse, si nëna e saj. Nga ana tjetër, ekziston një shans i barabartë 50% (1 në 2) që vajza të trashëgojë një kopje normale të gjenit, në të cilin rast ajo do të jetë e shëndetshme dhe jo bartëse (Figura 3).
Figura 3: Si transmetohen sëmundjet recesive të lidhura me X nga bartëset femra

Figura 4: Si transmetohen sëmundjet recesive të lidhura me X nga burrat e prekur

Nëse një burrë me një sëmundje të lidhur me X ka një vajzë, ai gjithmonë do t'ia kalojë asaj kopjen e ndryshuar të gjenit. Kjo për shkak se meshkujt kanë vetëm një kromozom X dhe gjithmonë ua kalojnë atë vajzave të tyre. Kështu, të gjitha vajzat e tij do të jenë bartëse (Fig. 4). Si rregull, vajzat janë të shëndetshme, por rrezikohen të kenë djem të sëmurë.
Nëse një burrë me një sëmundje të lidhur me X ka një djalë, ai kurrë nuk do t'ia kalojë atij kopjen e ndryshuar të gjenit. Kjo për faktin se meshkujt ua kalojnë gjithmonë kromozomin Y djemve të tyre (nëse kalojnë kromozomin X, do të kenë një vajzë). Kështu, të gjithë djemtë e një njeriu me një sëmundje të lidhur me X do të jenë të shëndetshëm (Fig. 4).
Çfarë ndodh nëse pacienti është i pari në familje që diagnostikohet me këtë sëmundje?
Ndonjëherë një fëmijë me një çrregullim gjenetik të lidhur me X mund të jetë i pari në familje që diagnostikohet me këtë çrregullim. Kjo mund të shpjegohet me faktin se një mutacion (ndryshim) i ri në gjen ka ndodhur në spermën ose vezën nga e cila është zhvilluar fëmija. Në këtë rast, asnjë nga prindërit e fëmijës nuk do të jetë bartës i sëmundjes. Gjasat që këta prindër të kenë një fëmijë tjetër me të njëjtën sëmundje është shumë i ulët. Megjithatë, një fëmijë i sëmurë që ka një gjen të ndryshuar mund t'ua kalojë atë fëmijëve të tij në të ardhmen.
Testi i bartësit dhe diagnoza prenatale (testi gjatë shtatzënisë)
Për njerëzit që kanë një histori familjare të një çrregullimi recesiv të lidhur me X, ka disa opsione për testim. Një test bartës mund të kryhet tek gratë për të përcaktuar nëse ato janë bartëse të mutacioneve (ndryshimeve) në një gjen specifik në kromozomin X. Ky informacion mund të jetë i dobishëm kur planifikoni një shtatzëni. Për disa sëmundje të lidhura me X, mund të bëhet testimi prenatal (d.m.th., testimi gjatë shtatzënisë) për të përcaktuar nëse foshnja e ka trashëguar sëmundjen (për më shumë informacion, shihni broshurat e marrjes së mostrave të vileve korionike dhe amniocentezës).
Anëtarët e tjerë të familjes
Nëse dikush në familjen tuaj ka një sëmundje të lidhur me X ose është bartës, mund të dëshironi ta diskutoni këtë me anëtarët e tjerë të familjes suaj. Kjo do t'u japë grave në familjen tuaj mundësinë, nëse dëshirojnë, t'i nënshtrohen një analize (një analize të veçantë gjaku) për të përcaktuar nëse ato janë bartëse të sëmundjes. Ky informacion mund të jetë gjithashtu i rëndësishëm për të afërmit kur diagnostikojnë sëmundjen. Kjo mund të jetë veçanërisht e rëndësishme për ata të afërm që kanë ose do të kenë fëmijë.
Disa njerëz mund ta kenë të vështirë të diskutojnë gjendjen e tyre gjenetike me anëtarët e tjerë të familjes. Ata mund të kenë frikë nga shqetësimi i anëtarëve të familjes. Në disa familje, për shkak të kësaj, njerëzit përjetojnë vështirësi në komunikim dhe humbasin mirëkuptimin e ndërsjellë me të afërmit.
Mjekët gjenetikë zakonisht kanë përvojë të gjerë në trajtimin e këtyre llojeve të situatave familjare dhe mund t'ju ndihmojnë të diskutoni problemin me anëtarët e tjerë të familjes.
Ajo që është e rëndësishme të mbani mend
- Gratë që janë bartëse të një sëmundjeje të lidhur me X-në kanë një shans 50% për të kaluar një kopje të ndryshuar të gjenit tek fëmijët e tyre. Nëse një djalë trashëgon një kopje të modifikuar nga nëna e tij, ai do të jetë i sëmurë. Nëse një vajzë trashëgon një kopje të modifikuar nga nëna e saj, ajo do të jetë bartëse e sëmundjes, si nëna e saj.
- Një burrë me një çrregullim recesiv të lidhur me X do t'ia kalojë gjithmonë vajzës së tij kopjen e ndryshuar të gjenit dhe ajo do të jetë bartëse. Megjithatë, nëse është një çrregullim dominues i lidhur me X, atëherë vajza e tij do të preket. Një burrë kurrë nuk ia kalon kopjen e ndryshuar të gjenit djalit të tij.
- Një gjen i ndryshuar nuk mund të korrigjohet - ai mbetet i ndryshuar gjatë gjithë jetës.
- Gjeni i ndryshuar nuk është ngjitës, për shembull, bartësi i tij mund të jetë një dhurues gjaku.
- Njerëzit shpesh ndihen fajtorë që kanë një çrregullim gjenetik në familjen e tyre. Është e rëndësishme të mbani mend se ky nuk është faji i askujt apo rezultat i veprimeve të dikujt tjetër.